Curated Repository Of Well-resolved Non-covalent Interactions
C · R · O · W · N
153,005 protein–ligand complexes, rigorously preprocessed and energy-minimized. Bridging the gap between structural quality and chemical diversity for next-generation ML models.
Existing databases force a trade-off between quality and scale
Machine learning models for protein–ligand interactions need both structural reliability and broad chemical coverage. Current resources offer one or the other — never both.
Curated but narrow
Databases like PDBBind and HiQBind offer carefully checked structures but cover only a fraction of the PDB. With a few tens of thousands of entries each, they under-represent the true diversity of known protein–ligand interactions, limiting the generalization of models trained on them.
Broad but noisy
Large-scale resources like PLInder catalogue nearly 650,000 systems across the entire PDB, but apply minimal quality filtering. Unresolved atoms, steric clashes, incorrect bond assignments, and missing quality annotations introduce systematic noise into training data.
Affinity-centric bias
Many benchmarks are organized around experimentally measured binding affinities, which cover only a subset of structures and introduce heterogeneous assay conditions. This excludes thousands of informative structures for which no affinity data exist.
CROWN bridges this gap
A fully automated preprocessing pipeline retains PLInder's broad coverage while enforcing rigorous structural standards — including constrained energy minimization, a step absent from all other available datasets.
From 649,915 systems to 153,005 curated complexes
CROWN applies five quality filters interleaved with two structural processing stages, each targeting a specific aspect of structural reliability — from crystallographic resolution to post-minimization stability.
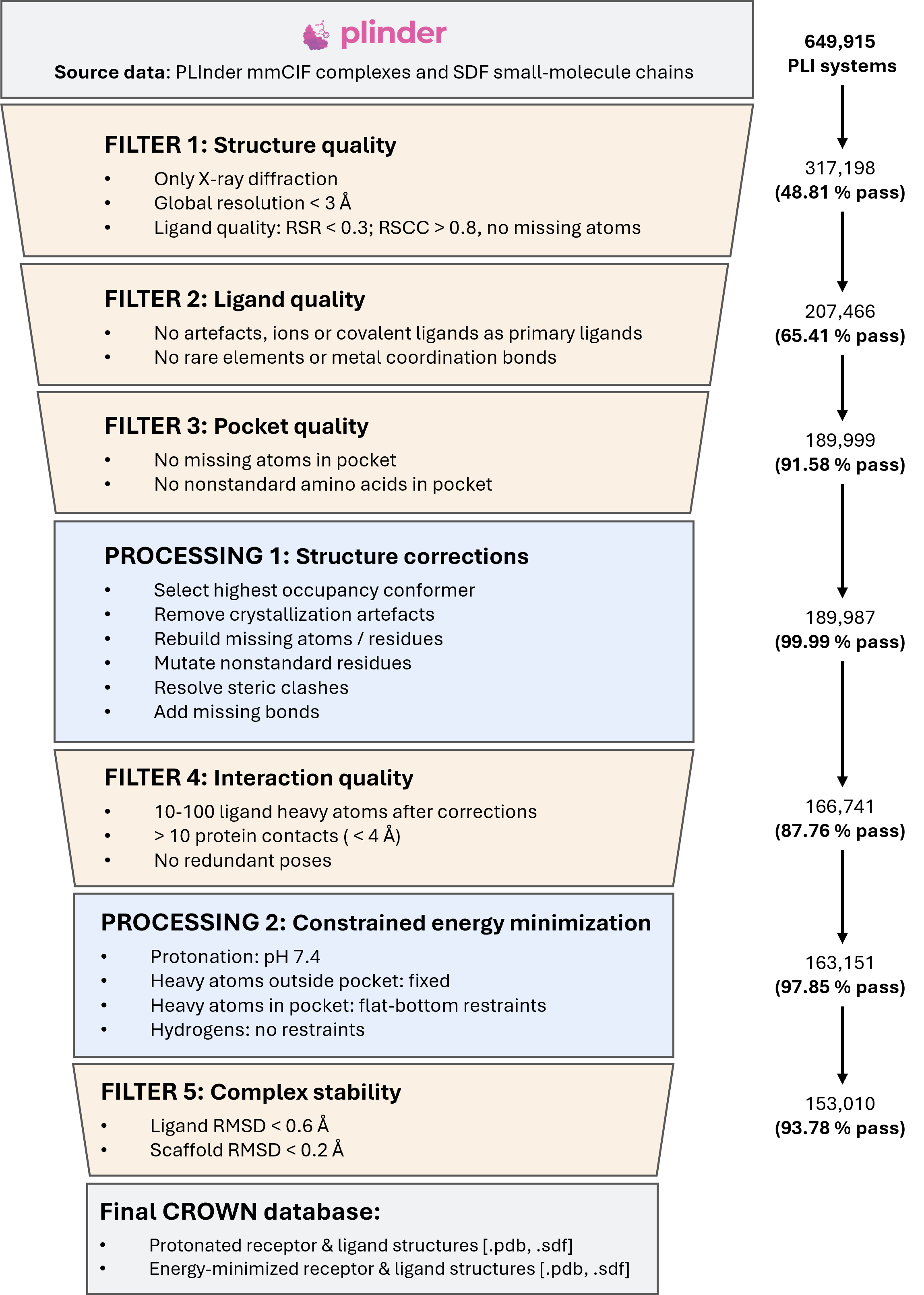
Preprocessing pipeline for CROWN, illustrated as a staged attrition funnel. Starting from 649,915 protein–ligand interaction systems sourced from PLInder, the pipeline applies five quality filters (beige) addressing structure, ligand, pocket, interaction, and stability quality, and two structural processing stages (blue) performing automated corrections and constrained energy minimization at pH 7.4.
What distinguishes CROWN
Every complex has been quality-filtered, structurally repaired, protonated at physiological pH, and energy-minimized with custom restraints — producing a structurally uniform collection ready for machine learning.
Verified electron density
Every entry has validated RSR (< 0.3) and RSCC (> 0.8) scores. No structures with missing quality annotations — a blind spot present in all other datasets, including PDBBind and HiQBind.
Drug-like, non-covalent only
Ions, crystallization artifacts, and covalently bound ligands are removed. Ligands contain 10–100 heavy atoms with > 10 protein contacts, ensuring only meaningfully engaged binding poses are retained.
Automated structural repair
Missing atoms rebuilt, alternate conformers resolved, steric clashes removed, and broken bonds repaired — all with a 99.99% success rate across nearly 190,000 systems.
Constrained energy minimization
A custom flat-bottomed tethering potential allows atoms to relax within crystallographic uncertainty (0.25 Å) while preserving experimental geometry. This step is unique to CROWN among all available datasets.
Geometry-centric philosophy
CROWN treats the 3D interaction geometry as the primary source of information — not binding affinities. This avoids affinity-label bias and includes thousands of structures with no reported Kd or IC50.
Broad chemical coverage
~4× more protein and species diversity than PDBBind or HiQBind, with 26,746 unique ligand types and 13,523 Murcko scaffolds — including PROTACs, macrocycles, and larger drug-like molecules.
CROWN in the landscape of protein–ligand databases
A side-by-side comparison of dataset scope, ligand diversity, and structural quality across five widely used resources.
| Property | PDBBind | HiQBind | BioLiP2 | PLInder | CROWN |
|---|---|---|---|---|---|
| Dataset scope | |||||
| Total entries | 19,449 | 31,573 | 86,458 | 649,915 | 153,005 |
| Unique PDB-CCD pairs | 17,758 | 17,247 | 22,720 | 201,836 | 64,502 |
| Unique PDB IDs | 17,758 | 17,088 | 17,701 | 111,867 | 55,208 |
| Unique UniProt IDs | 3,354 | 2,642 | 14,933 | 22,243 | 12,352 |
| Unique CATH IDs | 583 | 443 | 1,017 | 1,565 | 976 |
| Unique species | 861 | 715 | 3,466 | 4,882 | 3,209 |
| Affinity annotations | ✓ | ✓ | ✗ | ✗ | ✗ |
| Ligand diversity | |||||
| Unique CCD IDs | 13,956 | 12,428 | 6,519 | 47,300 | 26,746 |
| Unique Murcko scaffolds | 8,251 | 7,660 | 3,213 | 22,743 | 13,523 |
| Oligo ligands | 2,632 | 676 | 2,711 | 36,379 | 10,572 |
| Ion ligands | 0 | 0 | 1,805 | 22,728 | 0 |
| Covalent ligands | 870 | 24 | 1,708 | 32,276 | 0 |
| Artifact ligands | 15 | 34 | 523 | 18,626 | 0 |
| Structure issues | |||||
| Missing bonds | 990 | 114 | 2,104 | 37,422 | 0 |
| Steric overlaps | 323 | 313 | 656 | 5,670 | 0 |
| Unresolved ligand atoms | 463 | 1 | 1,564 | 18,815 | 0 |
| Unresolved pocket atoms | 1,102 | 955 | 1,579 | 12,457 | 0 |
| Non-standard pocket residues | 307 | 245 | 972 | 4,473 | 0 |
| Structure corrections | |||||
| Protonation | ✓ | ✓ | ✗ | ✗ | ✓ |
| Energy minimization | ✗ | ✗ | ✗ | ✗ | ✓ |
Comparison of dataset scope, ligand diversity, and structural quality filtering across five protein–ligand interaction databases. CROWN uniquely combines zero structure issues with both protonation and energy minimization. Values for CROWN are reported for the corrected structures.
Freely available under CC BY 4.0
Browse, search, and download individual entries or the complete dataset. The full preprocessing pipeline is open-source.